| *604384 | |||||||||||||||||||||||||||
| ATPase, Ca(2+)-TRANSPORTING, TYPE 2C, MEMBER 1; ATP2C1 | |||||||||||||||||||||||||||
| Alternative titles; symbols | |||||||||||||||||||||||||||
| ATPase, Ca(2+)-SEQUESTERING SECRETORY PATHWAY Ca(2+) ATPase 1; SPCA1 PMR1, RAT, HOMOLOG OF
|
|||||||||||||||||||||||||||
| HGNC Approved Gene Symbol: ATP2C1 | |||||||||||||||||||||||||||
| Cytogenetic location: 3q22.1 Genomic coordinates (GRCh37): 3:130,569,368 – 130,735,555 (from NCBI) | |||||||||||||||||||||||||||
| Gene Phenotype Relationships | |||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||
| TEXT | |||||||||||||||||||||||||||
| Cloning | |||||||||||||||||||||||||||
|
Hu et al. (2000) identified an EST corresponding to the ATP2C1 gene within a 1.3-Mb YAC/BAC contig spanning the region of chromosome 3q21-q24 deleted in a family with Hailey-Hailey disease (HHD; 169600). This EST had been annotated as homologous to a yeast gene encoding a calcium ATPase with a function predicted to be related to that of SERCA2 (ATP2A2;108740). Hu et al. (2000) isolated the full-length cDNA corresponding to the human EST. Similar to other Ca(2+) ATPase genes, the ATP2C1 gene encodes 2 alternatively spliced transcripts, ATP2C1a and ATP2C1b. These transcripts differed in their C termini (encoding amino acids 877 to the end), but had the same expression patterns in all tissues examined. ATP2C1a was predicted to encode 919 amino acids, and ATP2C1b was predicted to encode 888 amino acids. The protein encoded by ATP2C1 was highly homologous (97% identity) to rat Pmr1, which in turn is homologous to the yeast calcium pump Pmr1, but less homologous to other calcium pumps. ATP2C1 is highly expressed in human epidermal keratinocytes and at various levels in other human tissues. Patients with HHD are not known to have extracutaneous manifestations of the disease. Hu et al. (2000)found no differences in ATP2C1 mRNA levels between skin taken from the axilla and skin from the buttock (sites particularly prone vs resistant to blistering, respectively, in HHD patients) of one normal individual and little change in ATP2C1 mRNA levels in normal human epidermal keratinocytes cultured with glucocorticoid.
|
|||||||||||||||||||||||||||
| Gene Structure | |||||||||||||||||||||||||||
|
Hu et al. (2000) detected 27 exons in the ATP2C1 gene. Dobson-Stone et al. (2002) detected 28 translated exons.
|
|||||||||||||||||||||||||||
| Mapping | |||||||||||||||||||||||||||
|
Hu et al. (2000) mapped the ATP2C1 gene to chromosome 3q21-q24.
|
|||||||||||||||||||||||||||
| Gene Function | |||||||||||||||||||||||||||
|
Kourtis et al. (2012) demonstrated that heat stroke triggers pervasive necrotic cell death and neurodegeneration in C. elegans. Preconditioning of animals at a mildly elevated temperature strongly protected from heat-induced necrosis. The heat-shock transcription factor HSF1 (140580) and the small heat-shock protein HSP-16.1 mediate cytoprotection by preconditioning. HSP-16.1 localizes to the Golgi, where it functions with the calcium- and magnesium-transporting ATPase PMR1 to maintain calcium homeostasis under heat stroke. Preconditioning also suppresses cell death inflicted by diverse insults, and protects mammalian neurons from heat cytotoxicity. Kourtis et al. (2012) concluded that their findings revealed an evolutionarily conserved mechanism that defends against diverse necrotic stimuli. In mouse cortical neurons and striatal cells, Kourtis et al. (2012) found that overexpression of crystallin alpha-A (123580), which colocalizes with the Golgi marker alpha-mannosidase-II (154582) and the PMR1 ATPase, was sufficient to protect mammalian neurons from heat stroke-induced death, even in the absence of preconditioning. Heat stroke caused massive necrotic death and axonal degeneration in neurons expressing short hairpin RNAs against Pmr1, even after preconditioning.
|
|||||||||||||||||||||||||||
| Molecular Genetics | |||||||||||||||||||||||||||
|
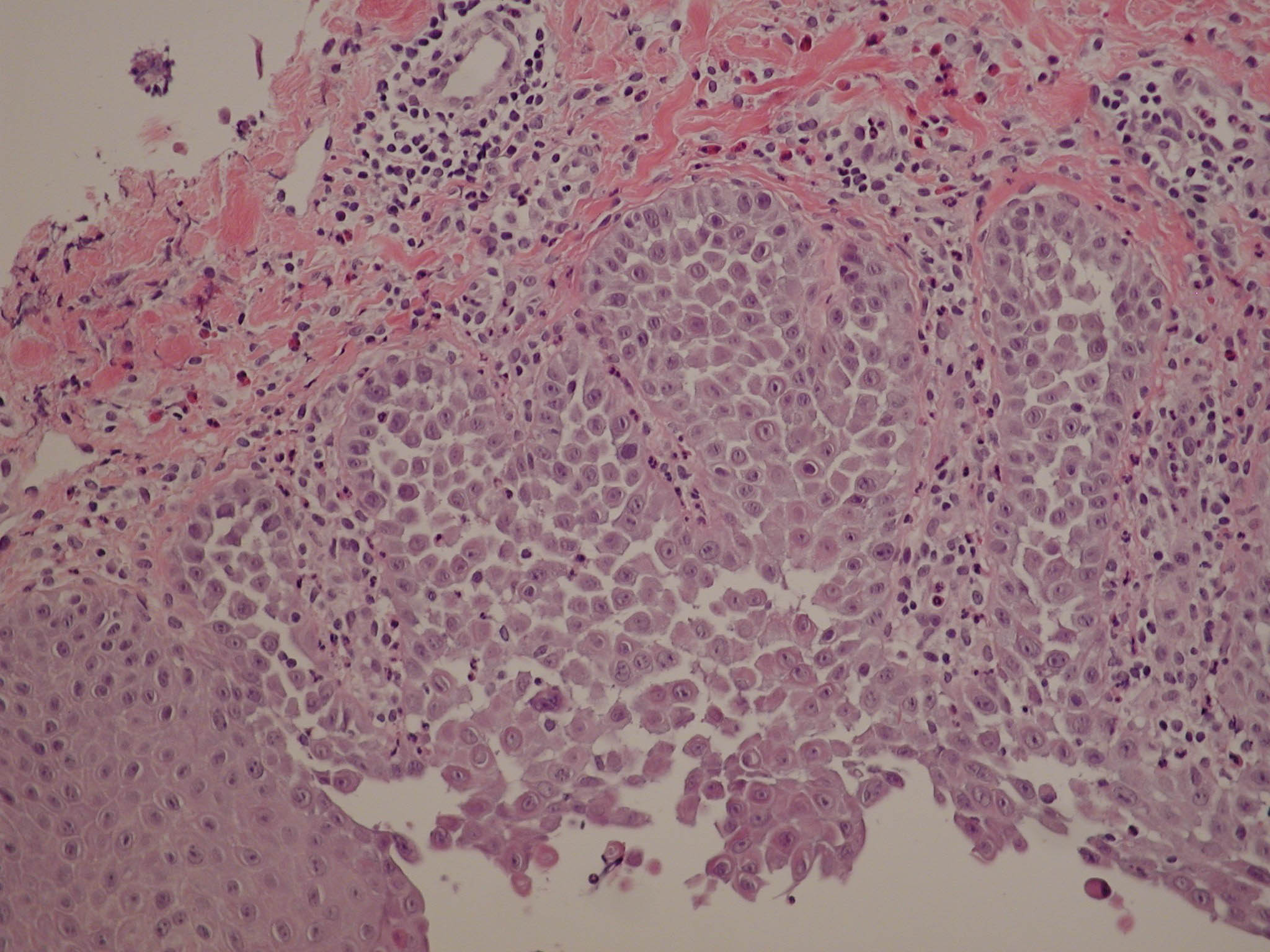
Hailey-Hailey disease (HHD; 169600) is an autosomal dominant disorder characterized by persistent blisters and erosions of the skin. By family linkage studies, the HHD region was localized to 3q21-q24. Study of a family carrying a deletion helped narrow the location. To screen HHD patients for ATP2C1 mutations, Hu et al. (2000) identified intron sites by comparison of genomic and cDNA sequences, designed primers flanking the 27 identified exons, and assessed PCR products from patients and controls by single-strand conformation polymorphism (SSCP) or conformation-sensitive gel electrophoresis (CSGE) analyses. Among 51 unrelated kindreds of European descent and 10 of Japanese descent, they identified 21 abnormalities (16/51 and 5/10). Of the abnormal sequences, 6 predicted single amino acid substitutions, 2 predicted aberrant splicing, and 13 predicted prematurely truncated products through frameshifts or single-basepair substitution. A high frequency of the last type of mutation supported a haploinsufficiency pathogenesis consistent with the complete deletion of the gene in 1 kindred and further suggested that calcium pumps of the PMR1 family function as monomers. The mechanism by which mutant ATPC1 causes acantholysis is unknown, but it may be through abnormally elevated cytoplasmic calcium or abnormally low Golgi Ca(2+) levels. Elevated cytoplasmic calcium might act by altering posttranslational modification of proteins or by inducing changes in gene expression.Sudbrak et al. (2000) identified 13 different mutations, including nonsense, frameshift insertion and deletions, splice-site mutations, and nonconservative missense mutations, in ATP2C1 in patients with Hailey-Hailey disease. The identification of ATP2A2 as the gene defective in Darier disease (124200) provided further evidence of the critical role of Ca(2+) signaling in maintaining epidermal integrity.Ikeda et al. (2001) reported ATP2C1 mutations in 11 Japanese patients with Hailey-Hailey disease. Some affected individuals had unique clinical features (generalization of Hailey-Hailey disease and generalized skin eruption resembling keratotic papules in Darier disease), but other affected individuals did not, suggesting the presence of intrafamilial phenotypic variations. These findings reinforced the conclusion that differences in clinical phenotypes in Hailey-Hailey disease are probably related to factors other than the type of causative mutation.
Chao et al. (2002) identified 7 different ATP2C1 mutations, 6 of them novel, in 7 Taiwanese kindreds with Hailey-Hailey disease. They found 3 deletion mutations, 2 nonsense mutations, 1 missense mutation, and 1 splicing mutation. Dobson-Stone et al. (2002) screened all 28 translated exons of ATP2C1 in 24 Hailey-Hailey disease families and 3 sporadic cases and identified 22 mutations (18 novel) in 25 probands. The novel mutations comprised 3 nonsense, 6 insertion/deletion, 3 splice site, and 6 missense mutations, and were distributed throughout the ATP2C1 gene. They noted that 6 of the mutations were found in multiple families in their study as well as in the studies of Sudbrak et al. (2000) and Hu et al. (2000). Haplotype analysis revealed that 2 of these were recurrent mutations. Comparison between genotype and phenotype in 23 families failed to yield any clear correlation between the nature of the mutation and clinical features of Hailey-Hailey disease. The extensive inter- and intrafamilial phenotypic variability suggested that modifying genes and/or environmental factors may greatly influence the clinical features of this disease. In a patient with unilateral segmental exacerbations of Hailey-Hailey disease, Poblete-Gutierrez et al. (2004) identified heterozygosity for a splice site mutation in exon 22 of the ATP2C1 gene (604384.0009). Haplotype analysis of the more severely affected segmental skin regions revealed consistent loss of the paternal wildtype allele, confirming the authors’ hypothesis that such segmental exacerbations represent a form of mosaicism with hemizygosity for the mutation. |
|||||||||||||||||||||||||||
| ALLELIC VARIANTS (Selected Examples): | |||||||||||||||||||||||||||
| Table View | |||||||||||||||||||||||||||
| ATP2C1, 4-BP INS, 767CCCT | |||||||||||||||||||||||||||
| In a family with Hailey-Hailey disease (169600), Hu et al. (2000) found a 4-bp insertion after nucleotide 767 in exon 10 of the ATP2C1 gene. The insertion resulted in a frameshift with a premature termination codon 42 amino acids downstream of the mutation. | |||||||||||||||||||||||||||
| ATP2C1, ALA304THR | |||||||||||||||||||||||||||
| In a family with Hailey-Hailey disease (169600), Hu et al. (2000) identified a G-to-T transversion of nucleotide 910 of the ATP2C1 gene, resulting in an ala304-to-thr amino acid substitution. | |||||||||||||||||||||||||||
| ATP2C1, ARG468TER | |||||||||||||||||||||||||||
| In a family with Hailey-Hailey disease (169600), Hu et al. (2000) found a 1402C-T transition in the ATP2C1 gene that altered codon 468 from arginine to stop. | |||||||||||||||||||||||||||
| ATP2C1, 4-BP DEL, 2374TTTG | |||||||||||||||||||||||||||
| In 2 unrelated families with Hailey-Hailey disease (169600), Hu et al. (2000) found an identical 4-bp deletion of 2374delTTTG in the ATP2C1 gene. The 2 families had different alleles of the D3S1587 marker, a locus less than 100 kb from the mutant gene, on the mutant chromosome. This may indicate that these were independent mutations. The deletion resulted in a premature termination codon 10 amino acids downstream of the mutation. | |||||||||||||||||||||||||||
| ATP2C1, IVS11, G-A, -1 | |||||||||||||||||||||||||||
| Sudbrak et al. (2000) identified a G-to-A transition at the 3-prime end of intron 11 (nucleotide position 852) of the ATP2C1 gene in a family with Hailey-Hailey disease (169600). The effect on the cDNA was not determined. | |||||||||||||||||||||||||||
| ATP2C1, CYS490PHE | |||||||||||||||||||||||||||
| In a Japanese patient who represented a sporadic occurrence of Hailey-Hailey disease (169600), Yokota et al. (2002) reported a cys490-to-phe (C490F) amino acid substitution that arose from a 1469G-T transversion in exon 17 of the ATP2C1 gene. | |||||||||||||||||||||||||||
| ATP2C1, 1-BP DEL, 2460G | |||||||||||||||||||||||||||
| In a Japanese patient who represented a sporadic occurrence of Hailey-Hailey disease (169600), Yokota et al. (2002) reported a frameshift mutation in the ATP2C1 gene, 2460delG, that resulted in a premature termination codon at exon 25. | |||||||||||||||||||||||||||
| ATP2C1, LEU584PRO | |||||||||||||||||||||||||||
| In a Japanese patient who represented a sporadic occurrence of Hailey-Hailey disease (169600), Yokota et al. (2002) reported a T-to-C transition at nucleotide 1751 in exon 19 of the ATP2C1 gene, resulting in a leu584-to-pro (L584P) amino acid substitution. | |||||||||||||||||||||||||||
| ATP2C1, IVS22, G-A, +1 | |||||||||||||||||||||||||||
| In a patient with unilateral segmental exacerbations of Hailey-Hailey disease (169600), originally reported by Vakilzadeh and Kolde (1985), Poblete-Gutierrez et al. (2004) identified heterozygosity for a G-to-A transition at the first base of the consensus splice donor site of exon 22 of the ATP2C1 gene. The mutation, which they designated 2146+1G-A, resulted in the skipping of the 69-bp exon 22. Haplotype analysis of the more severely affected segmental skin regions revealed consistent loss of the paternal wildtype allele, confirming the authors’ hypothesis that such segmental exacerbations represent a form of mosaicism with hemizygosity for the mutation. | |||||||||||||||||||||||||||
| REFERENCES | |||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||
|